Systemische Sklerose
oder Sklerodermie
Komplexe Erkrankung
mit vielfältigen Komplikationen
Herzlich willkommen im Sklerodermie Zentrum Hamburg
Die Philosophie des Sklerodermie Zentrums basiert auf dem täglichen Einsatz einer fachübergreifenden Medizin, die für die Diagnostik und Therapie der Sklerodermie-Patienten von eminenter Bedeutung ist. Dadurch erhalten Sie – unter Einsatz von Medizintechnik auf höchstem Niveau – eine präzise Diagnose und eine individualisierte Therapie.
Wir freuen uns auf Sie!
Was ist Sklerodermie (SSc)?
Die systemische Sklerose (SSc, Sklerodermie) zählt als „entzündlich-rheumatische Systemerkrankung” zur Gruppe der Kollagenosen und ist eine ernst zu nehmende und komplizierte Erkrankung. Der Name Sklerodermie leitet sich vom Griechischen ab und bedeutet übersetzt „harte Haut“ skleros (= hart) derma (= Haut). Anstatt des Begriffes „Sklerodermie“ wird heute vermehrt der Begriff „systemische Sklerose“ verwendet. Er verdeutlicht, dass die Verhärtung nicht nur das Organ „Haut“, sondern alle Körperorgane, z.B. auch die Blutgefäße und Lunge, betreffen kann.
Sklerodermie ist nicht heilbar, aber inzwischen behandelbar, so dass der Krankheitsverlauf verlangsamt und teilweise sogar gestoppt werden kann, also nicht voranschreitet.
Deswegen wird auf den Zusatz „progressive“ (= voranschreitende), wie er früher üblich war (Progressive Systemische Sklerose = SSC) heute verzichtet und einfach nur noch von einer systemischen Sklerose gesprochen.
Bei einigen Erkrankten sind die Veränderungen nur mit technischen Geräten messbar und werden vom Betroffenen gar nicht wahrgenommen. Andere Erkrankte sind deutlich am ganzen Körper betroffen und haben Verhärtungen im Gesicht und an den Fingern, die teilweise absterben. Auch kann die systemische Sklerose mit ernsthaften Beschwerden bei der Atmung, dem Schlucken und der Verdauung verbunden sein. Da die Krankheit sehr variantenreich ist, vergehen oft Monate oder Jahre, bis bei einem Patienten die richtige Diagnose gestellt werden kann.
Wie oft tritt Sklerodermie auf?
Mit zirka 40 Erkrankten pro eine Millionen Menschen gehört die systemische Sklerose zu den seltenen Erkrankungen. Es sind vier Mal mehr Frauen betroffen als Männer. Häufig tritt die Krankheit im 3. bis 5. Lebensjahrzehnt auf.
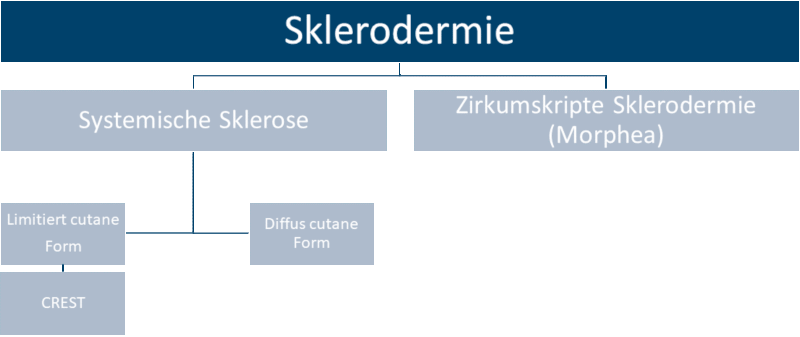
Klassifikation der Sklerodermie
Die internationale Klassifikation der systemischen Sklerose unterscheidet zwei Grundtypen:
- Limitiert cutane systemische Sklerose: Limitierte Form mit Hautbeteiligung distal der Handgelenke und des Gesichts bei fehlender oder späterer Beteiligung der inneren Organe. Eine Sonderform der limitierten SSc ist das sogenannte CREST-Syndrom, das folgende Symptome umfasst: Calcinosis cutis, Raynaud-Syndrom, Ösophagusbeteiligung, Sklerodaktylie und Teleangiektasien.
- Diffus cutane systemische Sklerose: Diffuse Form mit Hautfibrose proximal der Handgelenke und auch des Stamms mit früher Beteiligung der inneren Organe. Etwa 20 Prozent der Patienten werden dem diffusen Typ zugeordnet.
Krankheitsentstehung der Sklerodermie
Die systemische Sklerose ist von verschiedenen Verlaufsvarianten gekennzeichnet, die aber alle eines gemeinsam haben: Es kommt zu einer Verhärtung des Bindegewebes. Teilweise ist davon „nur“ die Haut betroffen (Sklerodermie), aber auch die inneren Organe und die Blutgefäße können verhärten und verengen (systemische Sklerose) und führen somit z.B. zu Lungenhochdruck (pulmonal arterieller Hypertonie, PAH). Eine weitere Folge der Gefäßverengung sind sehr schmerzhafte und die Lebensqualität einschränkende Geschwüre, meist an den Fingerspitzen (so genannte digitale Ulzerationen). Im Extremfall kann dabei das Gewebe so stark betroffen und entzündet sein, dass es abstirbt und eine Amputation notwendig ist.
Die genauen Ursachen der Sklerodermie sind nach wie vor unbekannt. Als Auslöser der Sklerodermie wird eine Fehlreaktion des Immunsystems (Autoimmunerkrankung) vermutet: Körpereigene Zellen (hier des Bindegewebes) werden als fremd oder fehlerhaft erkannt. Die Folge ist eine Entzündungsreaktion, bei der sich die Bindegewebszellen (Fibroblasten) übermäßig vermehren, so dass es zu einer Fibrose (Bindegewebszellenanhäufung) mit einer hohen Produktion an Kollagen kommt. Die Kollagenanhäufung führt zur Sklerose (Verhärtung) der Haut und zur Verengung von Blutgefäßen.
Es ist nach wie vor unklar, inwieweit zusätzlich genetische Faktoren oder Störungen der Bindegewebsneubildung bzw. der Gefäßregulation ursächlich sind. Auch andere Faktoren wie der Einfluss von viralen oder bakteriellen Antigenen, UV-Licht, Umweltgiften, Geschlechtshormonen, Medikamenten und bestimmten Tumoren könnten eine Rolle spielen.
Fest steht, dass bei der Entwicklung einer Sklerodermie folgende Gegebenheiten wichtig sind:
- Krankhafte Vermehrung des Bindegewebes (Fibrose)
- Schädigung der Blutgefäße (Vaskulopathie)
- Autoimmunerkrankung
- Genetische Veranlagung
- Vermehrte Bildung von Endothelin
Endothelin ist ein Stoff mit starker gefäßverengender Wirkung, der in der Innenauskleidung von Blutgefäßen (Endothel) gebildet wird. Endothelin spielt eine wichtige Rolle bei der Entstehung von Gefäßschäden: es verursacht Gefäßverengungen und trägt außerdem zur Entstehung von Fibrosen und Entzündungen bei. Aus verschiedenen wissenschaftlichen Untersuchungen ist bekannt, dass bei SSC-Patienten die Endothelin-Konzentrationen im Blut erhöht sind.